
|
Home |
Download |
Statistics |
Examples |
Help |
Contact |
Terms of Use |
 Navigation
Navigation1. Resource of Human Fusion Gene Breakpoints.
2. Clinical Assessment of each Gene in Pan-Disease Fusion Genes.
3. DNA sequence-based Fusion Gene Prediction and Fusion Genomic Features.
4. Open Reading Frame (ORF) Analysis and Generation of Fusion Sequences
5. Protein Functional Annotation.
6. Tumorigenic Mechanism of Action Scenarios of Fusion Genes
7. Personalized Fusion Protein Sequence Generation (FusionSeqDB)
8. Three Categories of Fusion Translation
9. Fusion Gene Targeting Studies
1. Resource of Human Fusion Gene Breakpoints.
We obtained RNA-seq based fusion gene breakpoint information from TCGA, CCLE, cBioPortal, ChimerDB4, ChimerKB4, ChildHoodFusions, and GTEx. From GenBank, we downloaded the nucleotide sequences and identified the breakpoints by running the BLAT. For the WGS-based fusions, we downloaded all the structural variant breakpoint information from dbVar. All genome coordinates of breakpoints were lifted over to hg19 since majority were identified from hg19.2. Clinical Assessment of each Gene in Pan-Disease Fusion Genes.
Individual genes can involve in the formation of fusion genes from multiple breakpoints with multiple partner genes in multiple cancer types. Based on these numerical features, we introduced a concept of the degree of frequency (DoF) score, which is the product of three values above each gene in our previous study. For example, in the application of this DoF to ~500 human kinomes, we identified multiple features of driver kinase fusion genes (Kim et al., 2016). We also introduced another metric, called the Major Active Isofusion Index (MAII) (Kim et al., 2017), which is calculated by dividing the number of observed samples with a particular fusion gene by its DoF score. Here, the isofusion refers to one particular gene fusion combination, with one particular partner gene and one particular breakpoint, in one particular cancer type. This new score (MAII) can give us the average frequency of each gene for each possible isofusion. By applying the MAII scoring to the transcription factor fusion genes, we found PLAU, which encodes plasminogen activator urokinase and serves as a biomarker for tumor invasion, was found to be consistently activated in the samples with the highest MAII scores. However, these trials are not utilizing all numerical features of individual genes in pan-cancer fusion genes. To better rank the importance of individual genes in pan-cancer fusion genes, we can utilize all existing features.To help choose the clinically significant kinase so that the cancer patients that have fusion genes can be better diagnosed, we need a metric to infer the assessment of kinases in pan-cancer fusion genes rather than relying on the sample frequency expressed fusion genes. Most of all, multiple studies assessed human kinases as the drug targets using multiple types of genomic and clinical information, but none used the kinase fusion genes in their study. The assessment studies of kinase without kinase fusion gene events can miss the effect of one of the mechanisms that enhance the kinase function in cancer. To fill this gap, we suggested a novel way of assessing genes using a network propagation approach to infer how likely individual kinases influence the kinase fusion gene network composed of ~5K kinase fusion gene pairs (Cheng et al., 2024). To select a better seed of propagation, we chose the top genes via dimensionality reduction like a principal component or latent layer information of six features of individual genes in pan-cancer fusion genes. We tried to infer the clinical assessment of genes in fusion gene network not only relying on the expressed sample size. In FusionGDB3, the users can access the # expressed samples, # expressed disease types, # partner genes, # breakpoints, DoF score, MAII score.
3. DNA sequence-based Fusion Gene Prediction and Fusion Genomic Features.
Previously, we developed a deep learning-based classifier between fusion gene and no fusion gene breakpoint sequences (FusionAI (Kim et al., iScience, 2021)). For all fusion genes whose breakpoints are located at the exon junction boundaries, we can run FusionAI. For all exon junction breakpoint fusion genes in three categories of ORFs (In-frame fusion genes, 5UTR-3CDS fusions (N-truncated proteins), and 5CDS-3UTR fusions (C-truncated proteins),) We ran the FusionAI and got the probability of fusion gene breakpoint.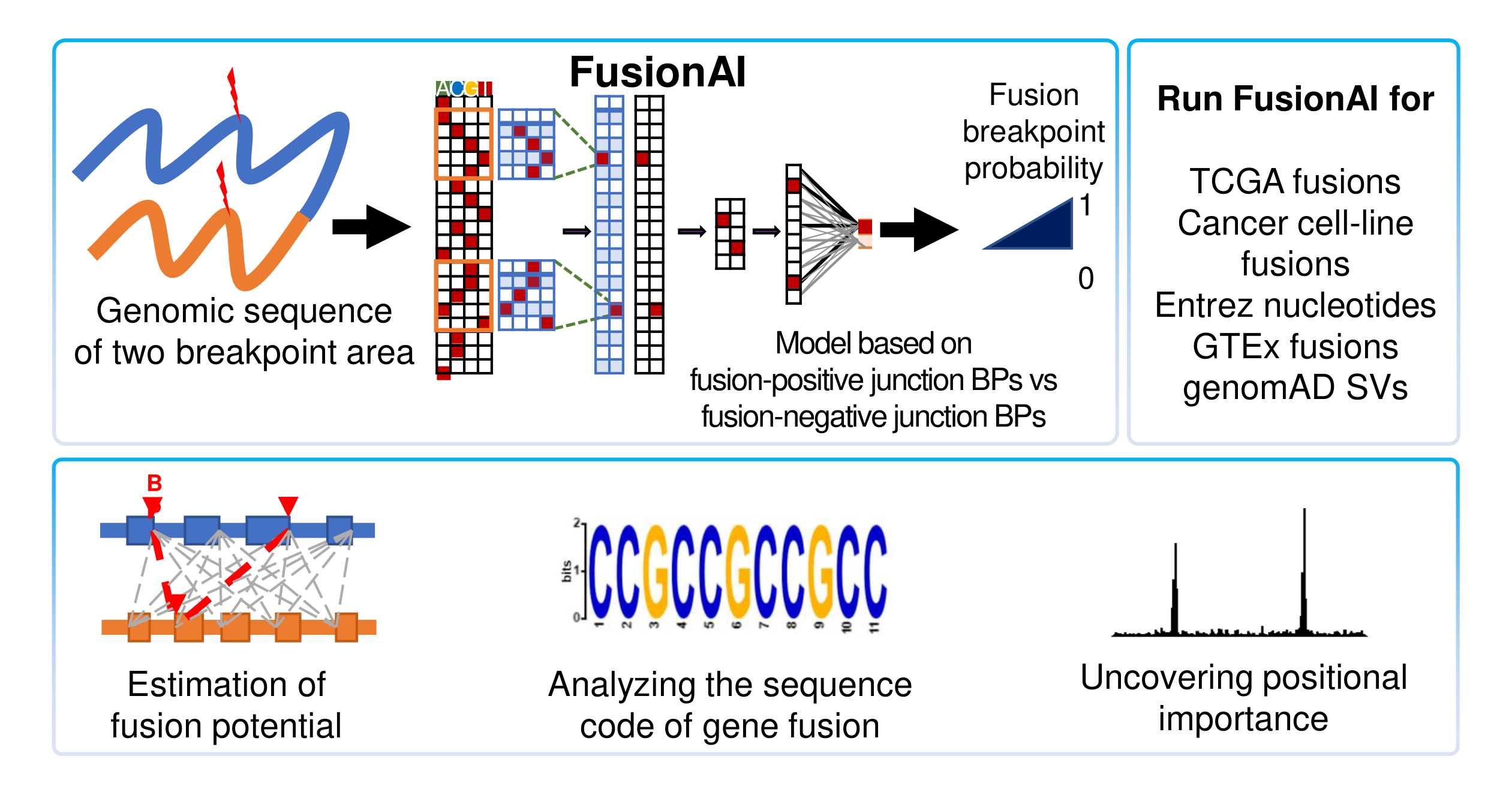
We explored the Epigenomic Profiling of human fusion gene breakpoint area using AlphaGenome. To explore the epigenetic baseline surrounding fusion junctions, we utilized AlphaGenome to profile the breakpoint area flanking +/- 5kb across multi-omics tracks: DNase-seq / ATAC-seq: Highlight chromatin openness and active regulatory regions. RNA-seq / CAGE: Capture transcript abundance, accumulation, and precise transcription start sites. ChIP-Histone / ChIP-TF: Map histone modifications and transcription factor binding architectures.
From this analysis of the epigenomic landscapes, we may be able to infer the Putative Functional Mechanisms. These baseline profiles do not definitively prove causality, but offer a data-driven framework to infer the putative mechanisms driving fusion oncogenesis.
a. Sustained Kinase Signaling (e.g., BCR-ABL1, EML4-ALK)Landscape: Shows highly stable and sustained RNA-seq signals across the transcript. This reflects continuous baseline transcription, supporting protein-level constitutive kinase activation.
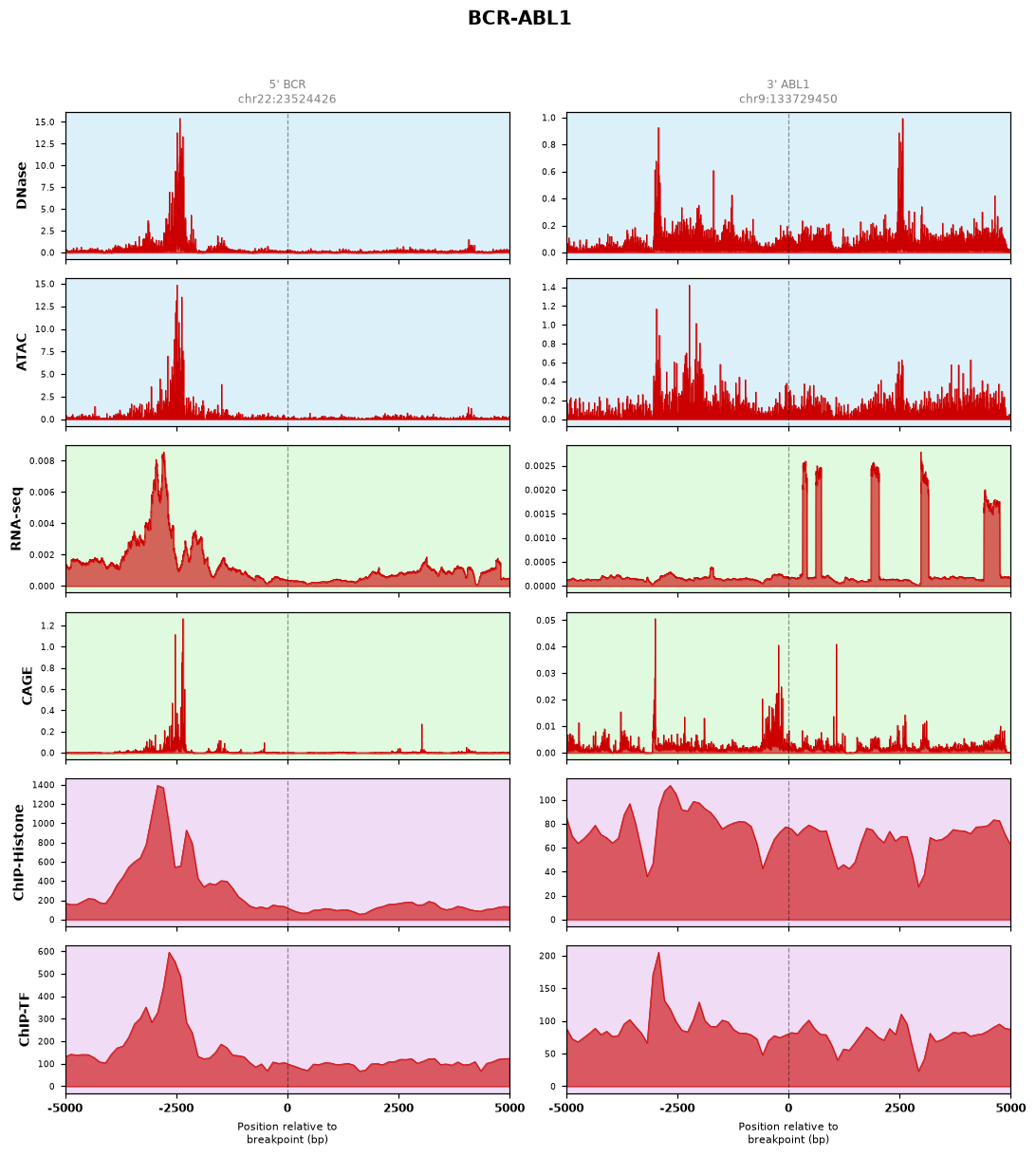
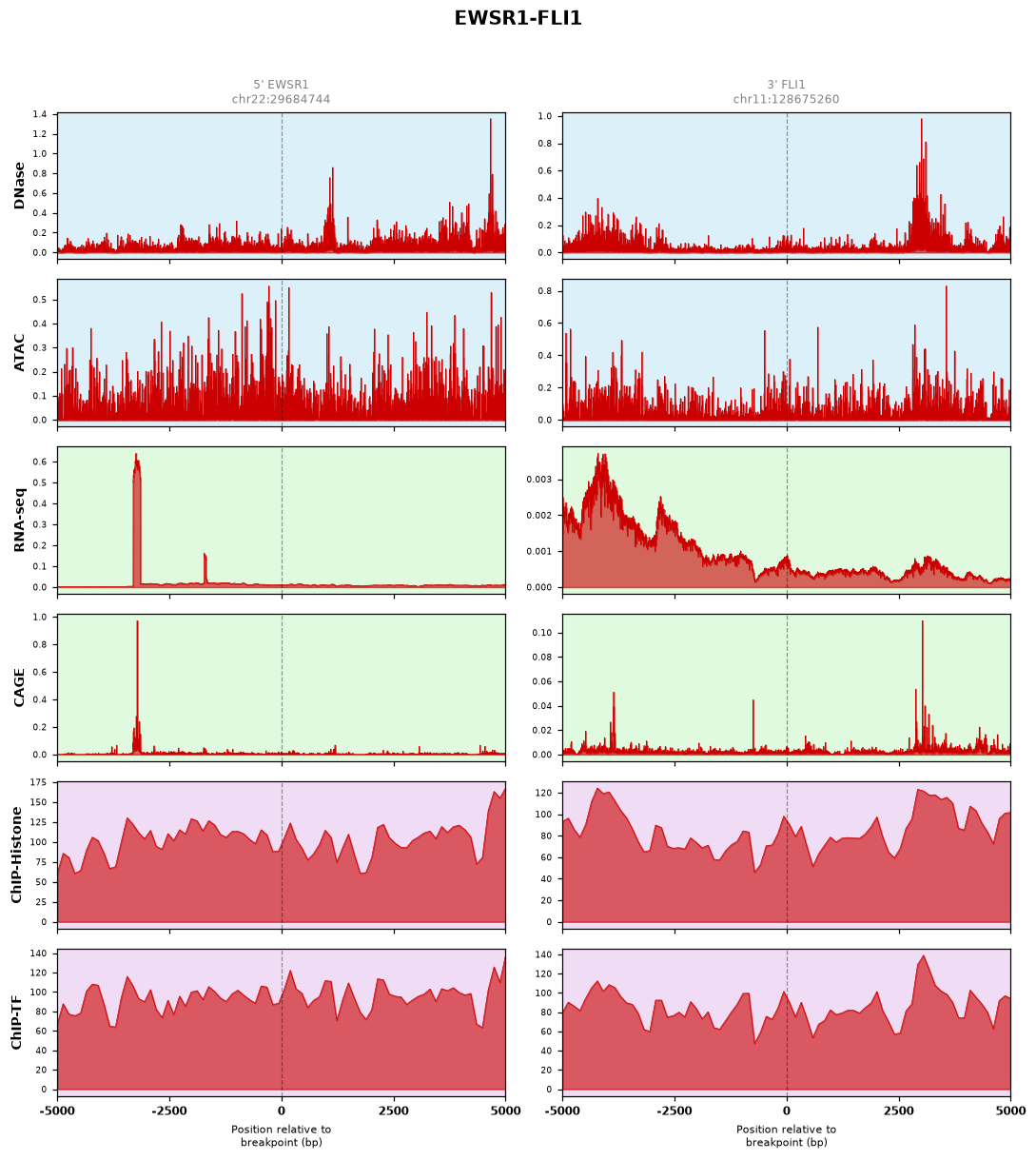
b. Active Transcriptional Hotspots (e.g., EWSR1-FLI1)Landscape: Features sharp, overlapping DNase/ATAC-seq peaks aligned with robust ChIP and CAGE signals near the breakpoint. This signature indicates a hyperactive chromatin state primed for aberrant transcription factor binding.
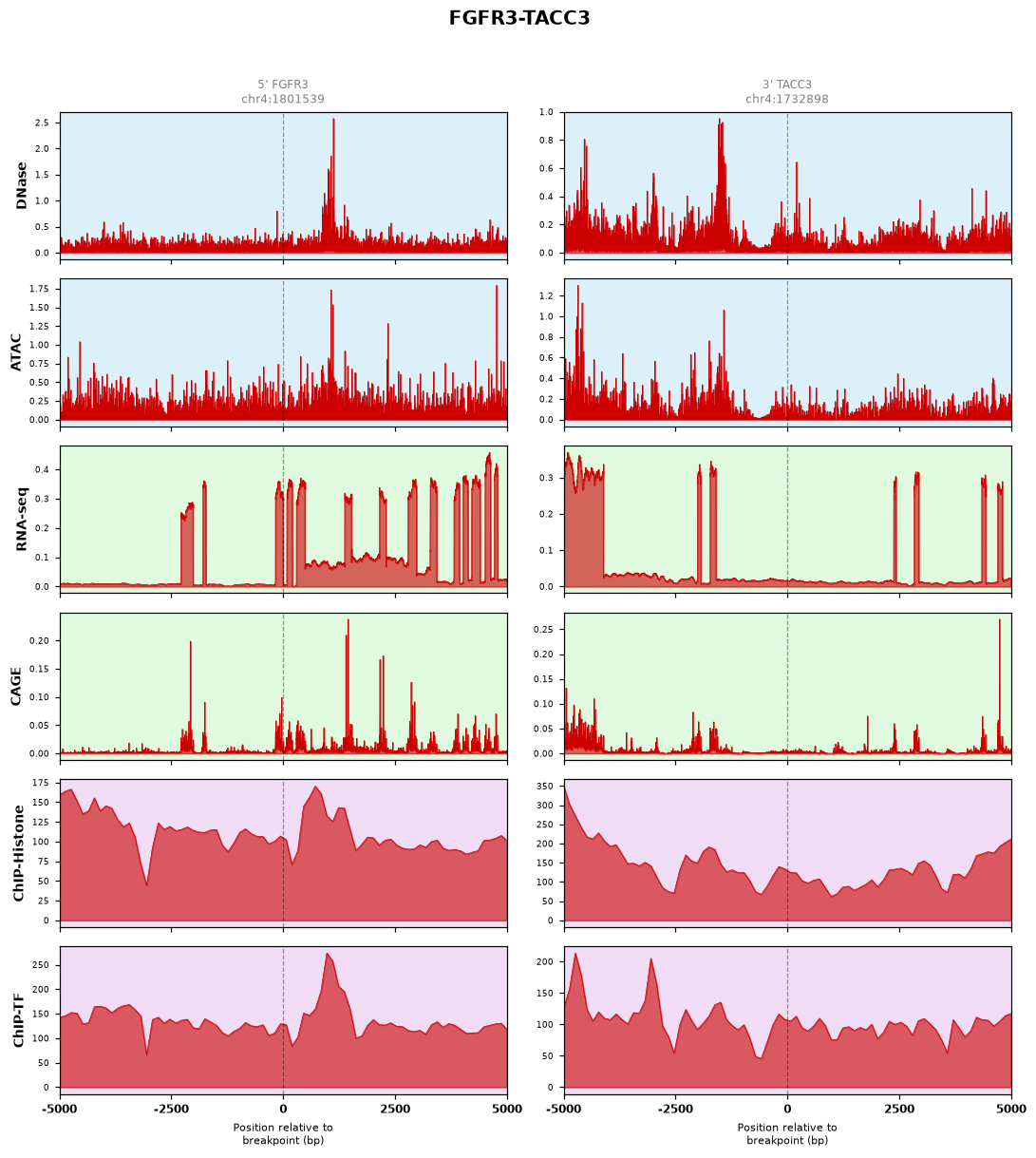
c. Potential Evasion of Post-Transcriptional Regulation (e.g., FGFR3-TACC3)Landscape: Characterized by a massive, continuous accumulation of RNA-seq signals upstream of the breakpoint, suggesting enhanced mRNA stability due to the loss of miRNA-mediated degradation (3'-UTR truncation).
d. Epigenetic Priming of Downstream Effectors (e.g., NUP214-ABL1)Landscape: Reveals strong DNase/ATAC-seq and ChIP peaks immediately preceding the breakpoint, followed by a sharp surge in RNA-seq signals, capturing the open chromatin environment that facilitates downstream expression.
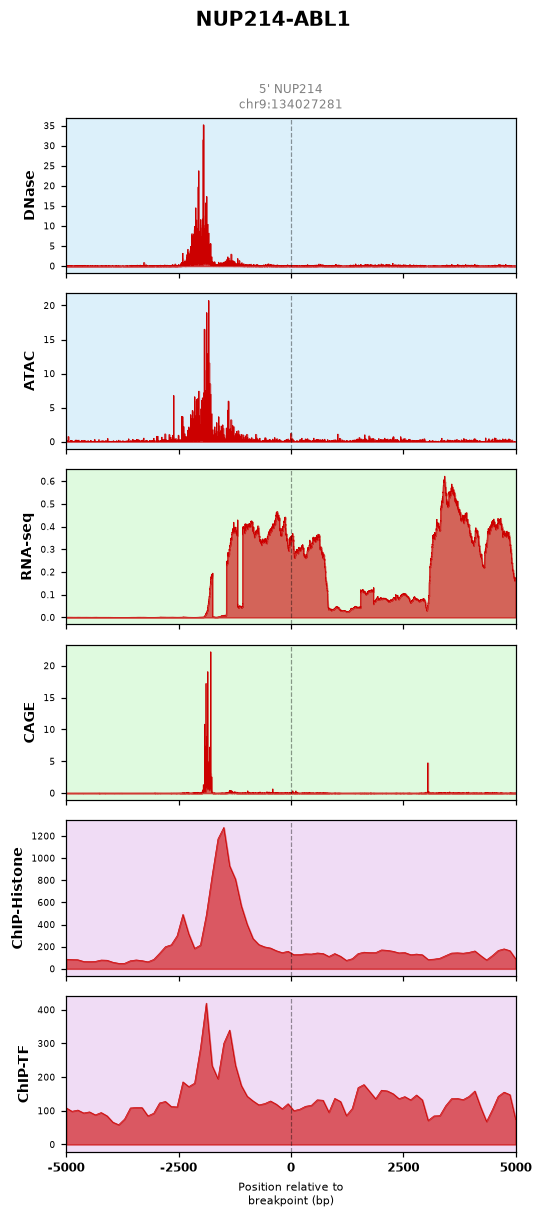
4. Open Reading Frame (ORF) Analysis and Generation of Fusion Sequences
To analyze the ORF fusion fusion genes, we provide three types of annotations. First, using in-house ORF annotation codes, we annotate the ORF of fusion transcript at given breakpoints and transcript isoforms. Between the 5’-partner gene and the 3’-partner gene, we checked the open reading frame of the full-length fusion transcript sequence. When both breakpoints of 5’- and 3’-genes are located inside of coding region (CDS) and the number of fusion transcript sequences from the transcription start site of 5’-gene to transcription end site of 3’-gene is a multiple of three, then we reported this fusion gene as ‘in-frame’. If there is one or two nucleotide insertion, then we reported as the ‘frame-shift’. Except for these two ORFs, there are 15 more ORFs such as ‘3UTR-CDS’, ‘3UTR-3UTR’, ‘3UTR-5UTR’, ‘3UTR-intron’, ‘CDS-3UTR’, ‘CDS-5UTR’, ‘CDS-intron’, ‘5UTR-CDS’, ‘5UTR-3UTR’, ‘5UTR-5UTR’, ‘5UTR-intron’, ‘intron-CDS’, ‘intron-3UTR’, ‘intron-5UTR’, and ‘intron-intron’. Second, we used ORFfiner. To make the input transcript sequences, for the in-frame fusion genes, 5UTR-3CDS fusion genes (N-truncated cases), 5CDS-3UTR fusions (C-truncated cases), we made full-length transcript sequences considering multiple gene isoforms and multiple breakpoints in the individual partner genes. Then, we input these fusion transcript sequences to the ORFfinder and chose the longest ORF for each fusion transcript as the fusion peptide sequence. Last, we used a deep learning classifier to distinguish coding vs non-coding transcripts, named as deepORF. For the deep learning model design, we adopted it from RNAsamba, a neural network-based assessment of the protein-coding potential of RNA sequences. By retraining the model using high-quality training data set as explained in the pipeline FugionGDB 2.0 paper, we could have dramatically increased the performance of the prediction of coding potentials. The prediction scores’ distributions using RNAsamba and deepORF are shown in Figure below.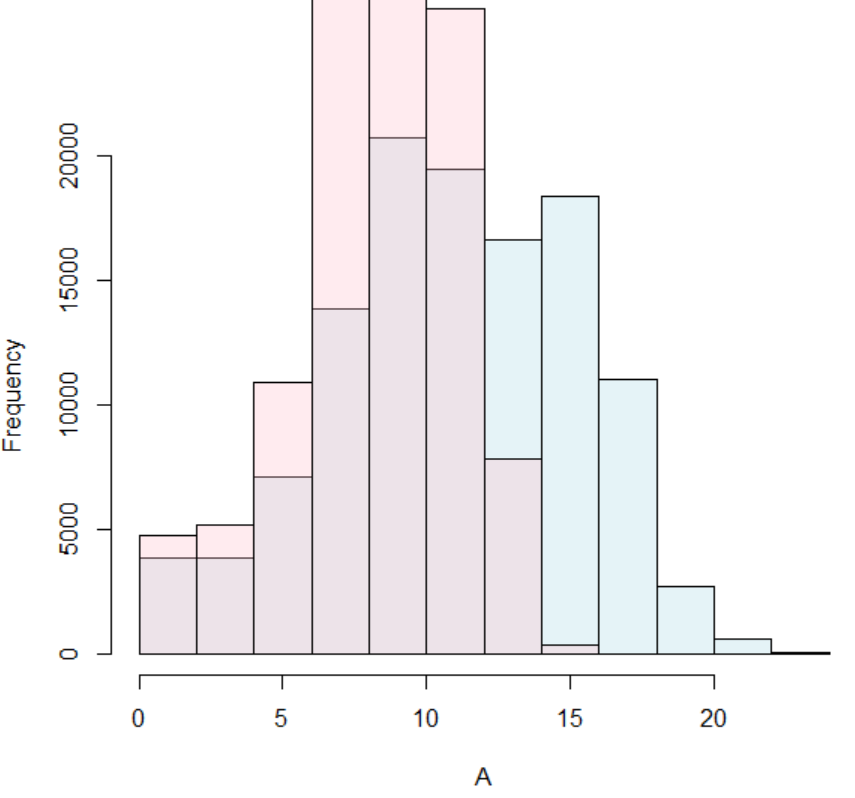
5. Protein Functional Annotation.
We searched the retention of 39 protein features of UniProt (six molecule processing features, 13 region features, four site features, six amino acid modification features, two natural variation features, five experimental info features, and 3 secondary structure features) at the fusion amino acid sequence level. Through this process, we also checked the retention of protein-protein interaction (PPI) at the fusion protein. Detailed information about all of the protein features is on the UniProt page.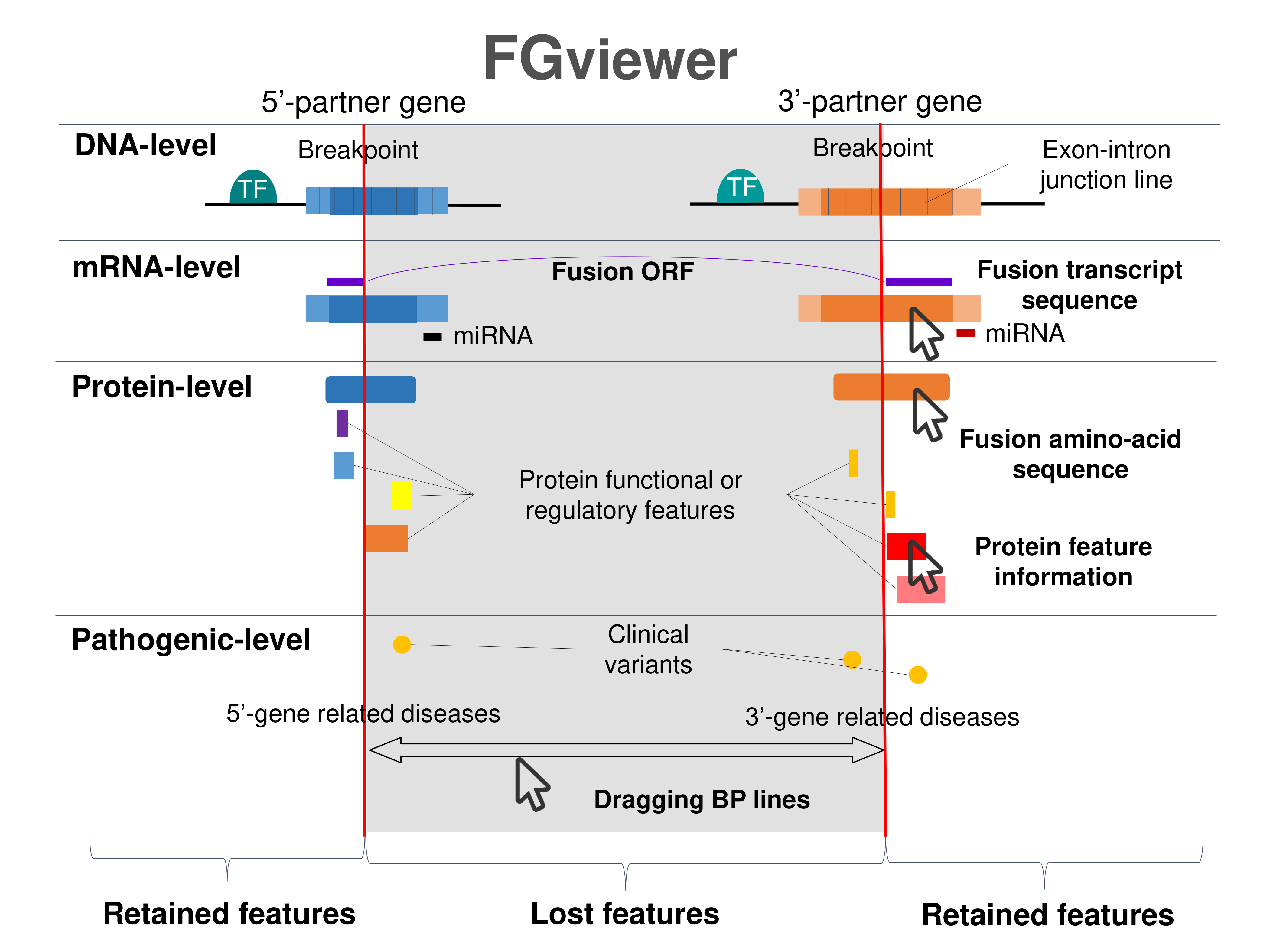
FGviewer (Kim et al., Nucleic Avids Research, 2020) provides functional feature annotations at four different levels: DNA-, RNA-, protein-, and pathogenic levels. The same breakpoint line across four tiers will classify between FG involving or non-involving zone with multiple types of functional features. For our fusion gene annotation page, we provide the protein-level annotation.
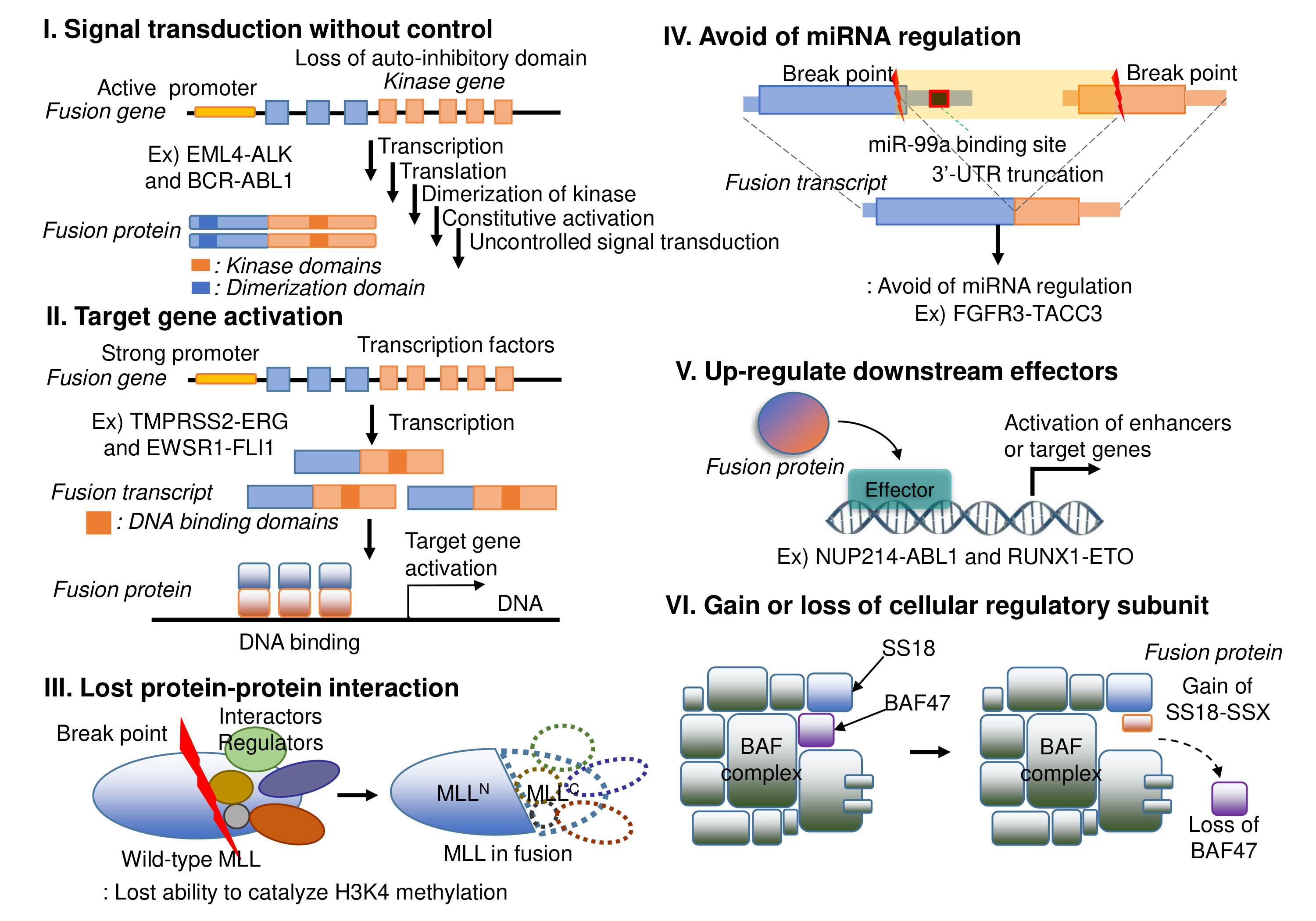
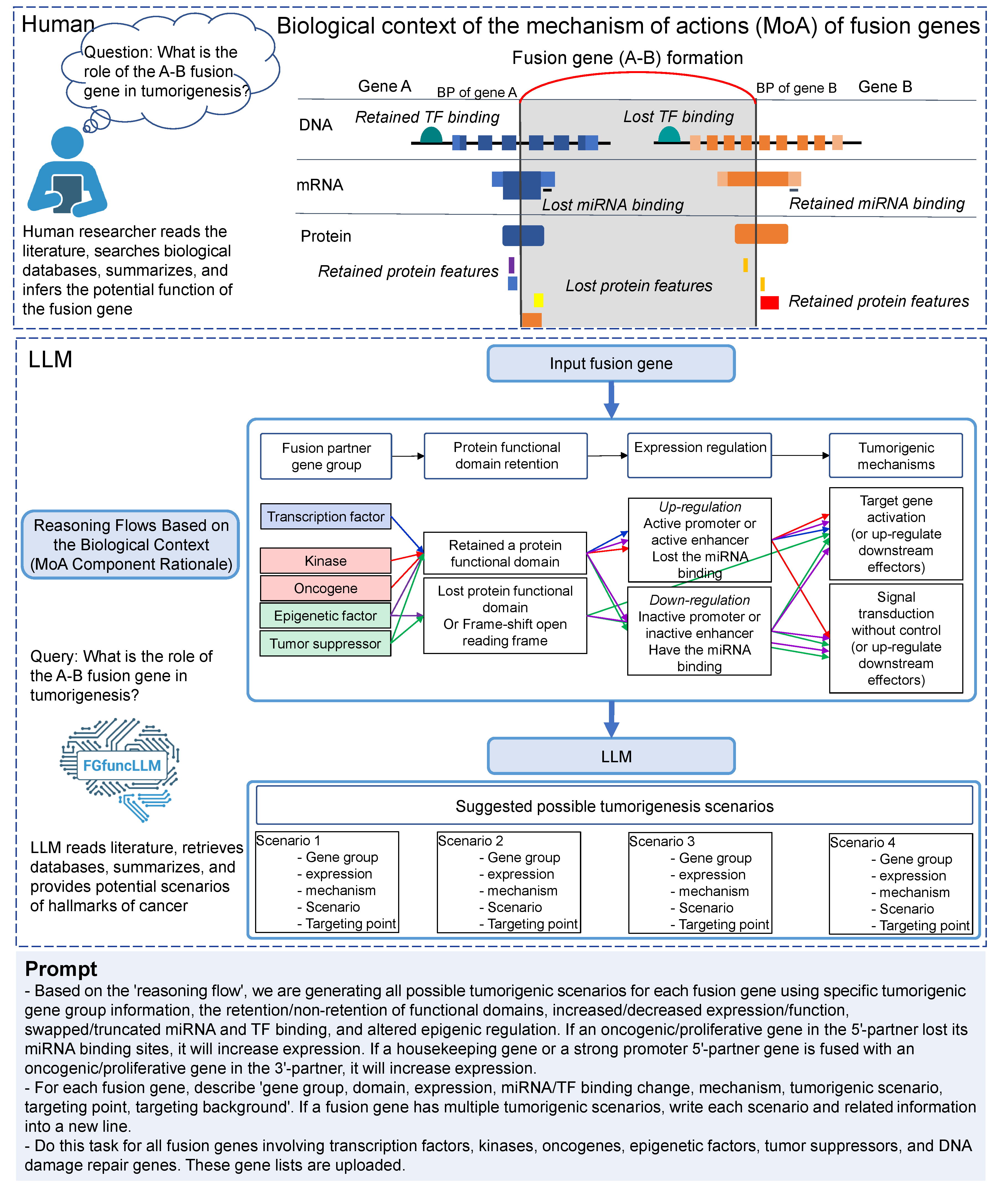
There are diverse categories of mechanisms of action of fusion genes in human diseases as above.
6. Tumorigenic Mechanism of Action Scenarios of Fusion Genes
Given the limited number of fusion genes studied (~1%), using a large language model (LLM) is a feasible approach to help researchers understand their functional roles. To maximize the potential of accurate functional annotation of LLM, human assistance is needed, such as providing a reasoning flow in a well-structured format, data based on the fluent biological knowledgebases. The major component of the fusion gene mechanism of action (MoA) is on the retained or lost protein functional features with abnormally regulated fusion gene expression, which are annotated in three levels of the central dogma of biology, like transcription factor swapping in DNA-level, miRNA regulation avoidance or swapping in RNA-level, and retained protein functional features in protein-level. We constructed a reasoning flow for the LLM to generate annotations of the potential mechanism of action and output the hallmark of cancer for the input fusion gene in five steps. For each step, we generated prompts with related biological knowledge from multiple reference resources.We developed a standalone Python pipeline to assign PubMed-supported literature evidence to Gemini-generated FusionGDB3 scenario annotations across four curated fields: mechanism, tumorigenic scenario, targeting point, and targeting background [1]. The input FusionGDB3 table was first parsed into claim-level units, with each row-column annotation treated as an individual claim-cell, followed by rule-based extraction of target genes and normalization of fusion-gene and claim text. For each claim-cell, the pipeline generated a tiered PubMed retrieval plan that prioritized fusion-specific evidence when biologically appropriate, followed by gene-specific evidence and template-generic evidence as fallback tiers. Candidate articles were retrieved via NCBI/PubMed E-utilities [2], after which GPT-5.4-mini was used as the default evidence judge, with GPT-5.4 as an upgrade model for selected cases [3]. To reduce the risk of fabricated references [4], the language model was isolated from the retrieval and PMID-generation steps: it was instructed to select PMIDs only from the retrieved candidate list, rather than generating PubMed records de novo, and all model outputs were subsequently validated against the candidate set before assignment. Each selected reference was labeled with a support scope, including fusion-specific, gene-specific, pathway-specific, template-generic, or no-reference support. The workflow also incorporated disk-based caching of PubMed and OpenAI responses, bounded concurrent query-unit evaluation to improve throughput, template-library overrides, and blocked-PMID rules for curated exceptions, high-reuse PMID auditing to identify overly generic references, and follow-up query rewriting/re-querying modules for unresolved, manually flagged, or high-reuse claim-cells. These post-processing and governance steps were designed to improve scalability while reducing weak, overgeneralized, or insufficiently specific literature assignments.
1. Team, Gemini, et al. "Gemini: a family of highly capable multimodal models." arXiv preprint arXiv:2312.11805 (2023).
2. Schuler, Gregory D., et al. "Entrez: Molecular biology database and retrieval system." Methods in enzymology. Vol. 266. Academic Press, 1996. 141-162.
3. OpenAI. Introducing gpt-5.4. https://openai.com/index/introducing-gpt-5-4, 2026.
4. Topaz, Maxim, et al. "Fabricated citations: an audit across 2· 5 million biomedical papers." The Lancet 407.10541 (2026): 1779-1781.
7. Personalized Fusion Protein Sequence Generation (FusionSeqDB)
So far, our fusion protein sequences have been made based on the reference genome sequences and these sequences are imported to UniProt. In this version, after analyzing the ORF, generation of the full-length fusion trancript seqeunces, and translation of 155K human fusion genes, we could generate more than 77K human fusion protein sequences. This time, to integrate the personalized variant context into the fusion protein sequences, we developed a computational pipeline and all combined fusion protein sequences into FusionSeqDB. For ~ 51,000 fusion genes in ~ 8,100 TCGA individual samples, we reflected the SNVs of individual genomes on the wild-type proteins separately, which are involved in fusion genes. We run the blastp for the reference-genome-based fusion protein sequences against both WT protein sequences and SNV/indel-reflected WT protein sequences. Then, we replace the fusion protein sequences with the matched SNVs/indels-reflected WT protein sequences. We reflected 500K (504,696) SNVs in 10K (10,018) TCGA patients, which were mapped to 6,446 unique fusion genes. For the CCLE data, we reflected 60K (60,296) SNVs in 1,920 cancer cell lines into 2,288 unique fusion genes (6,841 fusion transcripts with different breakpoints). From TCGA and CCLE, we generated 1.13 million (1,129,239) personalized fusion protein sequences.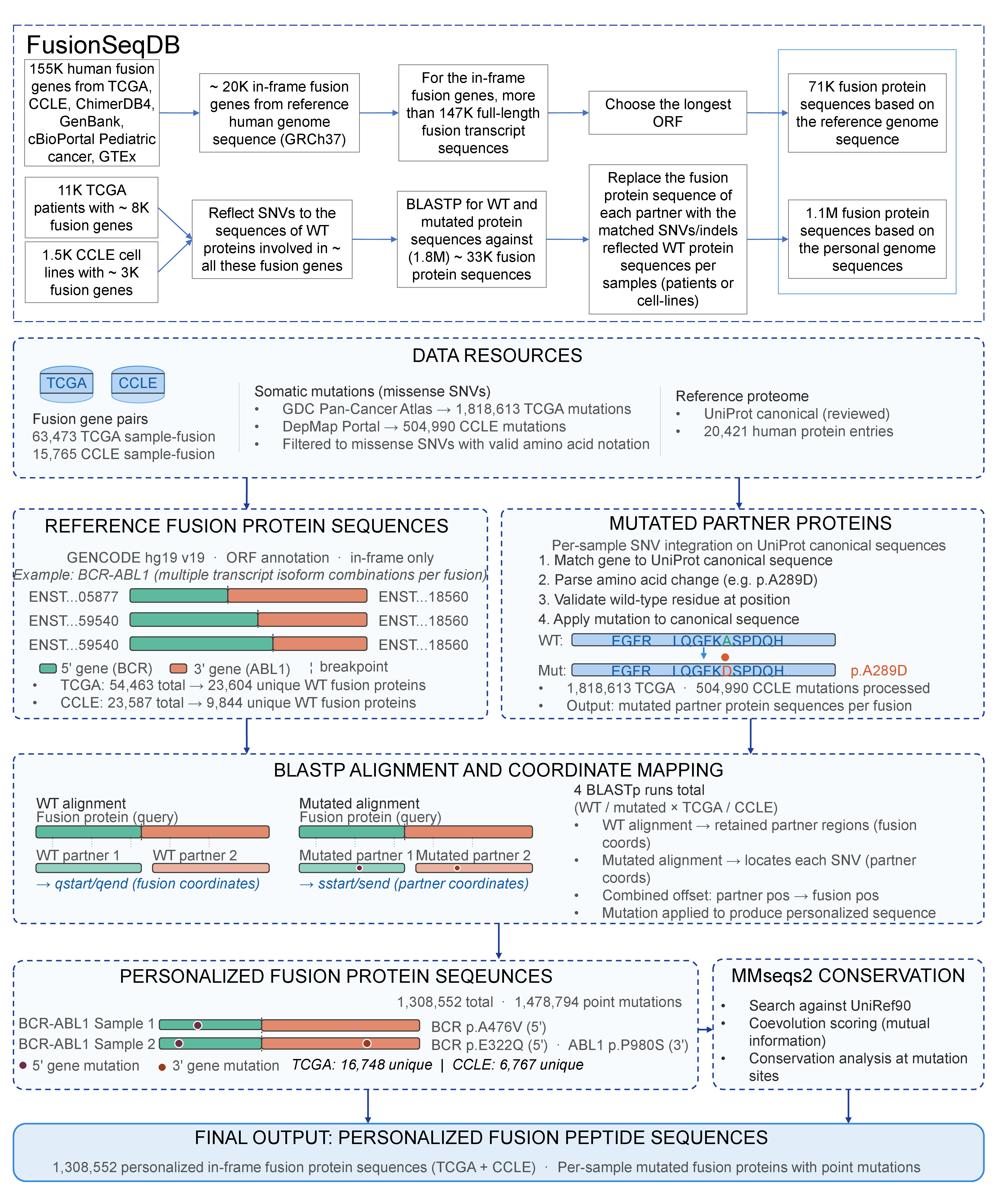
8. Three Categories of Fusion Translation
FusionGDB3 provides the information about three types of ORFs in fusion genes following the central dogma.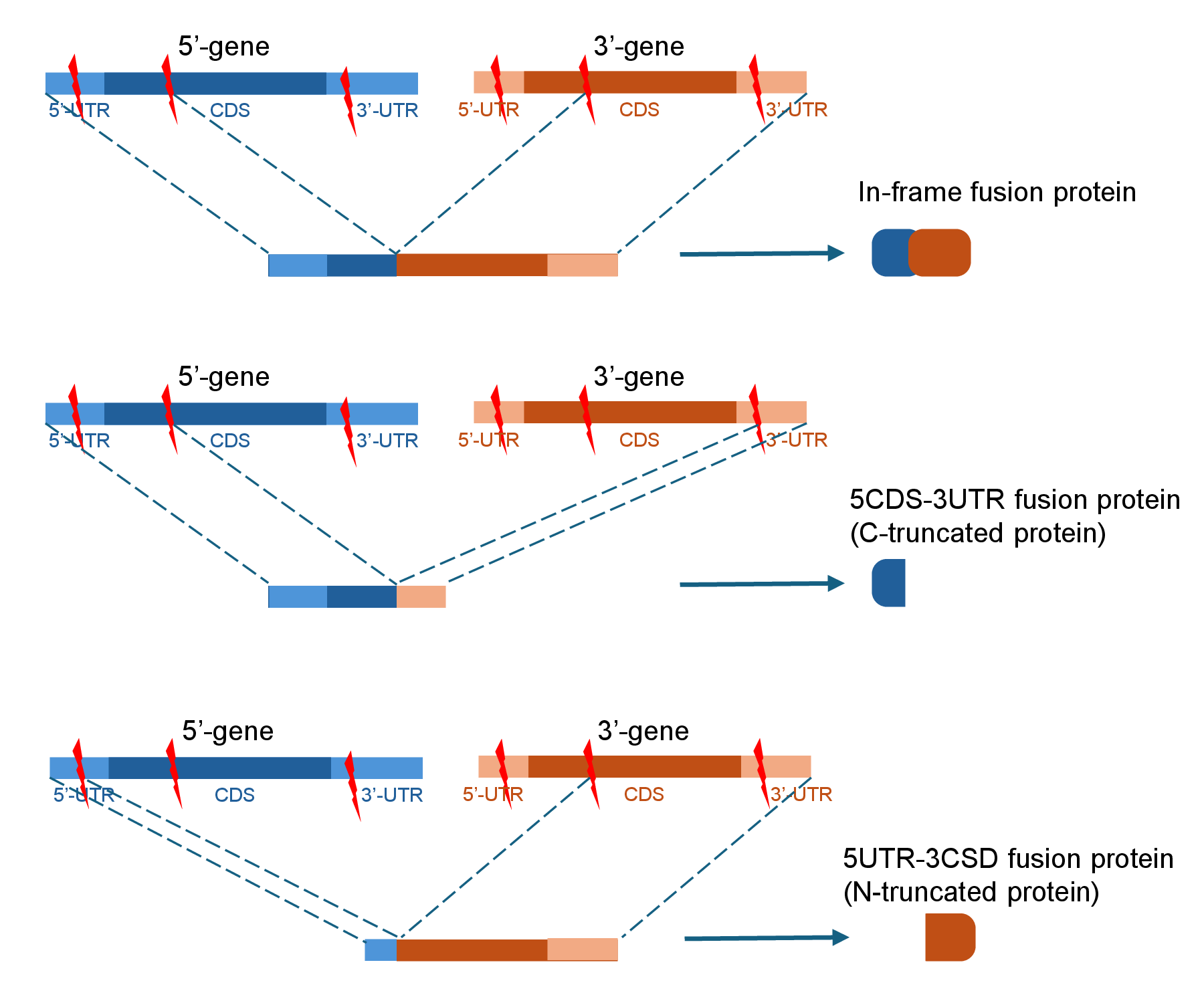
9. Fusion Gene Targeting Studies
To help experimentalTo advance the development of fusion protein targeting therapeutics, we need to know how fusion oncoprotein patients were/are treated in the clinic and what small molecules or therapies have been studied, which is critical for developing fusion protein targeting and personalized therapeutics. To fill this crucial gap, we developed FusionPub (Kumar et al., 2025). We performed the PubMed abstract search for 107K human fusion genes with all combinations of 14K drugs/small molecules (1.5 billion times). We also performed the PubMed abstract search for 18K human fusion proteins with all combinations of 8.5K and 8.3K gene synonyms of 5’- and 3’-partners with 14K drugs/small molecules (1.8 quintillion times). In FusionGDB3, the users can access to all fusion gene drug studies and more details can be accessed through FusionPub
 Copyright 2021-Present
Copyright 2021-PresentThe University of Texas Health Science Center at Houston (UTHealth)
Web File Viewing | Emergency Information
Campus Carry|Site Policies